医疗器械

IVDR简介
CE为法文CONFORMITE EUROPEENNE的首字母缩写,表示“欧洲统一”。
CE适用区域
欧盟EU+欧洲自由贸易联盟会员国,英国脱欧后,共有31个国家。
很多除欧盟外的国家,除美国FDA、日本PAL、澳大利亚TGA等,绝大数通行欧洲颁发的自由销售证书CFS。
| 法国 | 德国 | 英国(脱欧) | 爱尔兰 | 意大利 | 比利时 | 荷兰 | 西班牙 |
| 葡萄牙 | 卢森堡 | 瑞典 | 芬兰 | 奥地利 | 波兰 | 匈牙利 | 希腊 |
| 捷克 | 斯洛伐克 | 斯洛文尼亚 | 拉脱维亚 | 立陶宛 | 塞浦路斯 | 马耳他 | 罗马尼亚 |
| 丹麦 | 爱沙尼亚 | 保加利亚 | 冰岛 | 挪威 | 瑞士 | 土耳其 | 克罗地亚 |
CE标志是一种安全认证标志,凡贴有CE标志的产品均可在欧盟各成员国内销售,无须符合各个成员国的要求。使用CE标志,实现了商品在欧盟成员国范围内的自由流通,因此CE标志被视为制造商打开并进入欧洲市场的通行证。
在欧盟市场“CE”标志属强制性认证标志,不论是欧盟内部企业生产的产品,还是其他国家生产的产品,要想在欧盟市场上自由流通,就必须加贴“CE”标志,以表明产品符合欧盟《技术协调与标准化新方法》法规的基本要求, 加贴“CE”标志必须识别很多协调标准,这是欧盟法律对产品提出的一种强制性要求。
欧盟 医疗器械法规
| 名称 | 法规 | 发布日期 | 强制实施日期 |
| 医疗器械法规 | 2017/745,MDR | 2017-4-5 | 自2020-5-26起 |
| 体外诊断器械法规 | 2017/746,IVDR | 2017-4-5 | 自2022-5-26起 |
本次新发布的IVDR,在法规层级上从原先的Directive(指令)上升为Regulation(法规),标志着欧盟当局对医疗设备领域监管的进一步重视,同时也预示着在欧盟各成员国内医疗器械监管的尺度将得到进一步的统一。
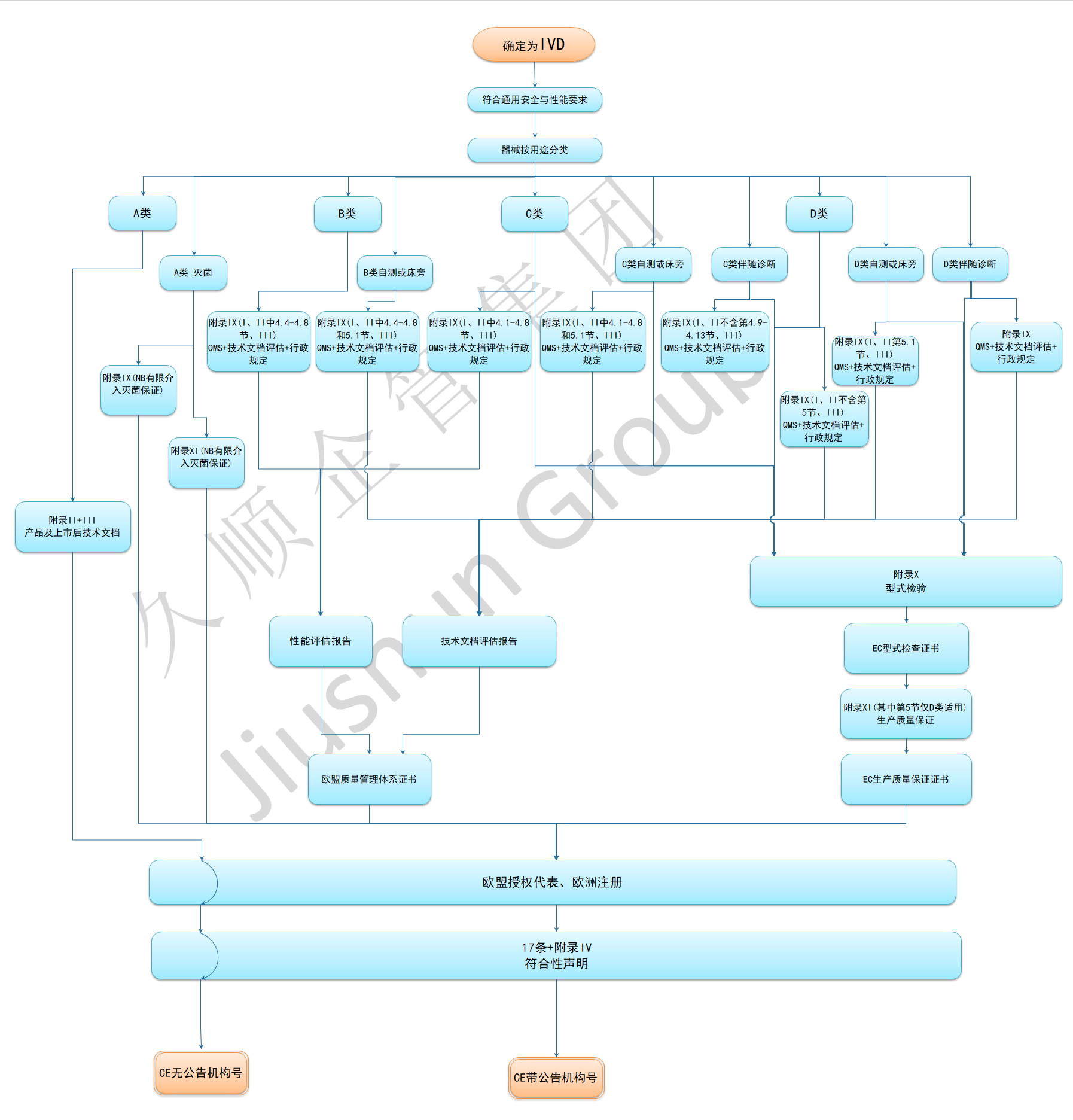
在IVDR法规中,对于体外诊断设备的监管依然是基于分类监管这个大框架,但是分类规则较原先的IVDD却发生了根本性的变化,即在IVDR中根据2017/746 法规第 V 章对IVDR医疗器械的符合性评估程序所做要求,基于产品的风险将所有的体外诊断设备由低到高分为了A、B、C、D四类,A(无菌) B,C,D类器械的认证需要公告机构进行评估,A类不需要公告机构的介入。
新法规IVDR与IVDD相比变化很大,为避免由于法规切换而造成对现有医疗系统的冲击,实现法规的“软着陆”,IVDR法规从生效(Entry into Force)到实施(Application)期间有5年的过渡期。IVDR法规的生效日为2017年5月25日,强制实施日为2022年5月26日。
在强制实施日期前依照旧法规IVDD颁发的CE证书,在证书有效期内持续有效;
在强制实施日期后依照旧法规IVDD颁发的CE证书,在实施日后的2 年失效。
体外诊断医疗器械(in vitro diagnostic device)是指通过对取自人体的标本进行体外检查的方式来提供医疗信息的医疗器械,包括制造商指定用于体外检查从人体取得的样本,包括血液及组织供体,无论单独使用或是组合使用的任何医疗器械,可包含试剂、试剂产品、校准材料、控制材料、成 套工具、仪表、装置、设备或系统,其唯一或主要目的是提供以下信息:
--生理学或病理学状态,或;
--先天畸形状况,或;
--确定安全性以及与可能接受治疗者的相容性;或
--检测治疗措施。
另外,样本容器也被认为是体外诊断医疗器械。样本容器是指其制造商明确规定主要用于盛装和保存从人体获得的样本以作体外诊断检查的器械,无论其是否为真空型。
但是,普通实验室用的产品不属于体外诊断医疗器械,除非此类产品根据其特性是制造商明确规定用于体外诊断检查用的。
体外诊断医疗器械CE认证的一般步骤:
步骤1. 分析该器械的特点,确定它所属的指令范围
步骤2. 确定该器械的分类(风险分级)
步骤3. 选择相应的符合性评价程序
步骤4. 选择公告机构
步骤5. 确认适用的基本要求/有关的协调标准
步骤6. 确认该器械满足基本要求/协调标准, 并使证据文件化
步骤7. 欧盟授权代表。
步骤8. 欧洲注册。
步骤9. 对于需要公告机构评审的器械,通过公告机构的符合性程序
步骤10. 起草符合性声明并加贴CE标志
IVDR CE技术文档的内容
1.目录表(版本状态,互相应用)。
2.生产者的名称和地址。
3.产品名称(所有的种类/型号)。
产品描述(如:预期用途,各种种类/型号的清单和描述,项目号、照片、图纸)。
4.器械样本的识别。
5.符合性声明。
6.其它文件,如证书,批准件。
7.适用标准清单。
8.基本要求检查表(附录1)。
9.风险管理。
10.产品历史(如产品的销售,投诉,产品更改)。
11.生产方式(生产流程图)。
12.质量控制过程(来科,过程中和最终控制)的描述,产品参数和批放行标准(原材料,元件,组件)。
13.如适用,与OEM生产者的协议。
14.过程验证;受控过程。
15.包装验证。
16.标签和使用说明书。
17.微生物状态的信息。
18.设计考虑(所有要求的总结)。
19.稳定性研究。
(单个元件/给件;稀释后/打开后;包括测试计划/测试标准)。
20.软件验证。
21.性能评估。
22.产品验证与确认:
23.产品临床试验报告,包括:
临床前评估:包含计划/执行/评估/报告/更新,临床前文献检索,实验室测试/模拟使用测试/计算机模拟/动物试验/与风险管理程序的接口,可用的临床前数据分析评价。
临床评估:包含临床评估计划/文献检索方法/文献检索出的文献/临床研究/验证/等效性/适用性/临床评估报告,上市后监管/PMCF计划/报告/与风险管理程序的接口,可用的临床数据分析评价。 临床使用概述及权威观点。临床评估报告。
以及针对含药器械、人体/动物来源组织或其衍生物制备的器械、引入人体并被吸收器械、具有测量功能器械等的相关附加信息。
附1.产品出厂检测报告。
附2.产品稳定性检测报告。
附3.基本要求检查表。
24.文献,出版物(技术,方法)。
25.自我测试器械所必须的特殊要求:
a.测试报告,包括非专业人员的研究
b.数据显示操作的适宜性
c.在标签和使用说明书上的信息
26.欧盟授权代表。
27.欧洲注册。
IVDR分类
IVDR 2017/746号法规附录VIII中详定7条规则,按医疗产品的风险程度,将产品分为Class A、Class B、Class C和Class D。
- 分类总则:
1. 分类规则的使用应基于器械的预期用途。
2. 若该器械将与其他器械共同使用,分类规则应分别适用于每种器械。
3. 对于在体外诊断医疗器械中的附件应与其配合使用的器械分开单独进行分类。
4. 驱动某一器械或影响器械使用的软件,应与该器械归为同一类别。
若该软件独立于任何其他器械,则按照其本身进行分类。
5. 预期与某一器械配合使用的校准品,应与该器械归为同一类别。
6. 赋值质控品包括定量和定性质控品,用于某一特定分析物或多种分析物的赋值质控品应与该器械归为同一类别。
7. 制造商应考虑所有分类和实施规则,便于为器械确定正确的分类级别。
8. 若制造商声称某个器械具有多种预期用途,使得该器械可归为多个类别,则它应被归入较高的类别。
9. 若多个分类规则适用于同一器械,产品类别以最高的为准。
10. 每条分类规则适用于一线试剂、确认试剂和补充试剂。
二.分类规则:
规则1.用于以下用途的器械归类为 D 类:
- 检测血液、血液成分、细胞、组织或器官,或其任何衍生物是否存在或显露传
染性因子,以评估它们是否适用于输血、器官移植或细胞给药。
- 检测是否存在或显露传染性因子,其会导致危及生命的疾病,并且具有高的或
可疑的传播风险。
-确定危及生命的疾病的病原体载量,其监控对于患者管理的过程十分关键。
示例:检测HIV/HCV/HBV/HTLV感染的试剂产品。
规则2.器械预期用于血型分型或组织分型,以确保用于输血或移植或细胞给药的血液、血
液成分、细胞、组织或器官具有免疫相容性,此类器械归类为 C 类,但用于确定以
下任何标记物的器械除外:
- ABO 系统 [A(ABO1)、B(ABO2)、AB(ABO 3)];
- 恒河猴(Rhesus)系统 [RH1(D)、RHW1、RH2(C)、RH3(E)、RH4(C)、
RH5(E)];
- KELL 系统 [Kel1(K)];
- KIDD 系统 [JK1(JKA)、JK2(JKB)];
- DUFFY 系统 [FY1(FYA)、FY2(FYB)],
在这种情况下,它们被归为 D 类。
示例:HLA分型系统
规则3.具有以下预期用途的器械为C类:
(a) 用于检测是否存在或显性传播病原体的;
(b) 用于检测是否在脑脊液或血液中存在某种高的或可疑的高传播风险的病原体;
(c) 用于检测病原体的存在,其报告结果若错误可带来引起个人、胎儿、胚胎或个
体的后代死亡或严重残疾的重大风险;
(d) 用于女性的产前筛查,确定其对感染原的免疫状况;
(e) 用于确定感染性疾病的状态或免疫状态,若其报告结果将会引起患者治疗决定
导致危及患者或患者后代生命的风险;
(f) 用作伴随诊断;
(g) 用于疾病分期,若其报告结果错误将会引起患者治疗决定导致危及患者或患者
后代生命风险的;
(h) 用于癌症的筛查、诊断或分期;
(i) 用于人类基因检测;
(j) 用于检测药用产品、物质或生物组分的水平,若其报告结果错误将会引起患者
治疗决定导致危及患者后代生命的风险;
(k) 对危及生命的疾病或病症患者,进行患者管理;
(l) 用于筛查胚胎或胎儿的先天性疾病;
(m) 用于新生儿的先天性疾病筛查,未能检测和治疗这些疾病可能导致危及生命的
情况或严重残疾。
示例:用于女性产前风疹或弓形体病免疫状态测试、凝血时间测试、唐氏筛查的产品。
规则4. (a) 自测器械归为 C 类,但用于妊娠检测、生育力测试、确定胆固醇浓度,以及测定尿液中糖分、红细胞、白细胞等器械除外,这些器械归为 B 类。
(b) 用于床旁检测器械,根据其本身特性单独分类。
示例:血糖监测类产品为C类,怀孕自测、生育能力测试、尿液测试条为B类。
规则5.以下器械归为 A 类:
(a) 一般实验室使用的产品、没有危险特征的附件、缓冲液、洗涤液、一般培养基
和组织染色液;
(b) 制造商专门用于体外诊断过程的器械;
(c) 样品容器。
示例:选择性/差异性微生物培养基,IVD仪器,平口尿液杯。
规则6.上述分类规则未涵盖的器械归类为 B 类。
示例:维生素、酶、激素之类的生理标记检测,代谢标记检测,脂泻病标记检测。
规则7.不具有定量或定性赋值的质控品的器械归类为 B 类。
IVDR申请流程