


一、监管框架
众所周知的是,无论处于哪种风险等级,所有医疗器械都需要将临床评价作为其技术文档的组成部分。
以眼镜产品为例,根据MDR的监管框架,处方眼镜与非处方眼镜均被作为医疗器械管理。而太阳眼镜则被视作个人装备,意味着制造商应根据MDR第 61条和附件XIV的part A策划、实施和记录临床评价。
二、眼镜类医疗器械的共性特征
眼镜通常被认为是一项成熟的技术(WET)(见MDCG2020-6),此类医疗器械的共同特点有:
·相对简单、常见且稳定的设计,基本无演变;
·其通用器械组具有众所周知的安全性,并且在过往与安全问题无关联;
·临床性能特征及其通用器械组采用护理器械的标准,在适应症和最新技术方面基本无变化;
·在市场上具有悠久的历史。
三、眼镜类医疗器械的临床评价过程
■ 一般来说,眼镜不需要临床试验或PMCF研究,因为此类产品基于成熟的技术,基本无创新,并且有着悠久的市场历史。可使用临床前和主动收集的上市后监督(PMS)数据以证明眼镜符合通用安全和性能要求(GSPR)。
■ 此外,制造商必须进行文献检索,包括类似器械的SOTA。SOTA的文献综述是关于器械的最新技术、临床背景、可替代和目前的技术,包括相关的安全和性能问题。
对类似器械开展潜在的安全和性能问题调查,并确认参数,此类参数用于确定所评价器械预期目的下收益风险比的可接受性。
■ 临床评价的第一步是策划行动,必须确定相关数据并随后进行评估。
■ 下一步是分析临床数据,并撰写临床评估报告(CER)。
临床评价过程如下图所示↓
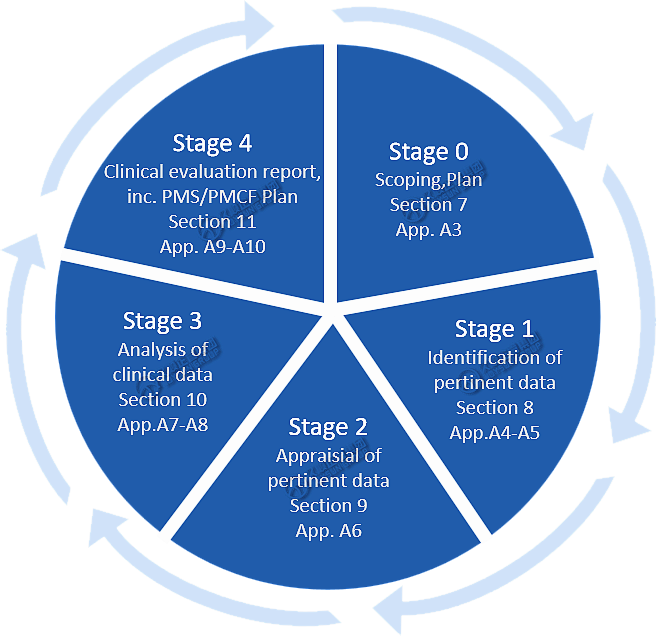
阶段0:范围、计划
阶段1:相关数据的识别
阶段2:相关数据的评估
阶段3:临床资料的分析
阶段4:临床评价报告、PMS/PMCF计划
临床前数据包括各领域的测试结果,如:生物相容性、生物力学、动物试验等。医疗器械的风险等级越低,应显示的临床前证据量就越大。
临床数据可能包括来自科学文献的数据以及PMS数据。
四、眼镜类医疗器械临床评价的挑战与应对
最常见挑战是临床评价的及时规划。由于CER是技术文档的最后一份文件,这意味着应事先提供其他文件。因此,编写 CER 前,应完成所有临床前测试和 PMS 数据的主动收集。
制造商应详尽测试其器械,以证明其符合标准(协调)和通用规范(如适用)。此外,应及时实施PMS数据的主动收集,并编制技术文档。
自身缺乏眼镜临床数据通常是制造商向MDR过渡的主要问题。然而,由于这类的许多器械在长时间内基本保持不变,因此来自市场经验的数据可支持成熟的技术(WET)方法。
充分记录的临床评估计划,是支持法规提交和CE标记过程的关键因素。
→欧盟CE注册:久顺企管值得您信赖与选择!始创于1996年,荷兰\西班牙\英国\美国\中国均设公司,近20年资深欧代、近30年合规技术专家,一站式快捷合规服务:CE注册取证\MDR和IVDR技术文档编写\合规策略\体系辅导\欧盟授权代表\上市后监督咨询等。
>>已成功布局欧盟临床试验渠道,提供欧盟临床试验一站式CRO服务:临床方案设计\临床试验方案的撰写\与当地实验室/医院合作,安排客户试验产品合规开展临床试验\包括但不限于:收集&整理&分析试验原始数据并出具临床试验报告。

