


一、欧盟警戒系统的概念
定义:涉及医疗器械事故(Incidents)和现场安全纠正措施(FSCA)的通知和评估系统。
目的:各欧盟成员国之间直接、尽早且统一地实施现场安全纠正措施(FSCA)。
即:通过收集医疗器械相关的上市后事故、不良事件的信息,在适当的情况下分发或传播此类信息,以防止不良事件再次发生。
相关指南:MEDDEV 2.12-1rev 8 GUIDELINES ON A MEDICAL DEVICES VIGILANCE SYSTEM医疗器械警戒系统指南
二、是否上报警戒系统的判断
如果医疗器械的特性和性能下降或使用说明书中的任何不足,可能导致或已导致患者\使用者的死亡或健康状况恶化,以及制造商出于安全原因将医疗器械撤出市场,制造商或欧盟授权代表有义务通知主管机构。
■ 必须上报主管机构的情况
必须同时满足以下三个条件:
1.事件已经发生;
2.正常使用医疗器械情况下发生的严重事故;
3.发生严重事故,具体表现为:死亡、健康恶化、严重公共健康威胁等。
■ 无需上报主管机构的情况
1.使用前器械已存在缺陷;
2.因患者自身原因导致事故;
3.器械超过使用期限;
4.器械被不正常使用;
5.出现故障但已发出实时警报;
6.已预期会发生的副作用;
7.发生几率微小,且并未有死亡、健康恶化等危害发生。
三、上报警戒系统的流程
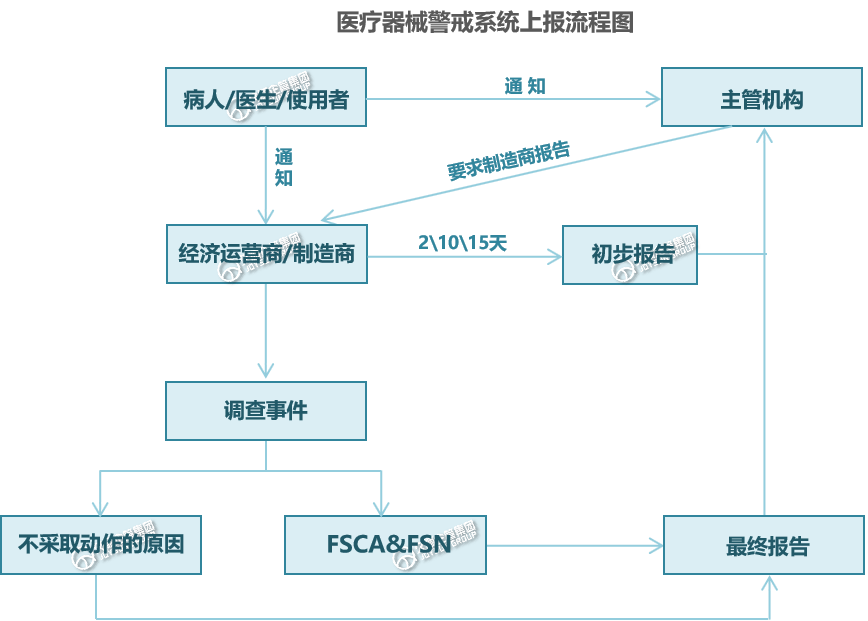
Step1. 事故报告将通过非专业人士、经销商、医疗保健专业人员、主管机构发送给制造商。
Step2. 收到事故报告后,制造商必须首先向主管机构发送初次报告,报告时限如下↓
☑ 严重公共健康威胁:2天;
☑ 死亡或无预期的严重健康恶化事件:10天;
☑ 其他严重事件:15天。
Step3. 制造商有义务针对所报告的事件开展调查;
Step4. 调查可能导致制造商进行事故风险评估,并通过现场安全通知(FSN)立即对使用者实施现场安全纠正措施(FSCA);
Step5. 制造商向主管机构提交最终报告,说明所采取纠正措施,并向使用者发出安全警报。
如果制造商未对事故报告采取任何措施,那么该制造商必须在提交给主管当局的最终报告中提供正当理由。
注意:所有报告均应以电子方式进行,上传至Eudamed系统,以面向公众开放。
警戒系统、上市后监督、经济运营商和器械登记等,都是CE所要求质量体系的重要组成部分。
如需体系建立、培训、升级等服务,可咨询[久顺企管集团]
久顺已为诸多制造商完成MDR/IVDR体系升级、公告机构体系监督审核中的MDR/IVDR发补意见,并已建立起完备的MDR\IVDR体系升级服务:GAP分析表、上市后监督系统、警戒系统等。
具体服务项目有:上市后监督计划PMS Plan、上市后性能/临床跟踪计划PMPF\PMCF Plan、趋势报告Trends Report、定期总结报告 Periodic Summary Report等记录报告的建立\执行\培训。

